Amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) are both devastating neurological diseases. ALS, also known as Lou Gehrig’s disease, progressively destroys nerve cells that control body movements, and affects as many as 30,000 Americans each year. FTD is the most common form of dementia diagnosed before the age of 60, impacts about 250,000 people in the US. There is no known cure to stop or reverse either condition, and only a handful of drugs approved by the FDA provide modest benefits for ALS patients, so the search for new therapies continues.
About 10% of ALS cases in the US are inherited, and up to 50% of FTD patients have a family history of FTD or related cognitive conditions. The two diseases often occur together as well: between 20 and 50% of ALS patients also develop FTD. In 2011, researchers based at Mayo Clinic discovered mutations in a human gene called C9orf72 that cause the inherited forms of both ALS and FTD. This was a crucial breakthrough, because it will help researchers understand how mutations in this gene cause neurodegeneration in both diseases.
To develop new therapies, the first necessary step is to generate an animal model, like a mouse that displays the same disease phenotype, or observable characteristics, of the human version of the disease. Then, scientists can use the model to dive deeper into the various cellular mechanisms and molecular processes underlying the disease. But developing an animal model for ALS and FTD has proven to be tricky because of the unusual nature of the human mutations found in the C9orf72 gene.
All genes contain segments of coding DNA, called “exons,” that carry instructions for encoding proteins. Most human diseases are caused by mutations in exons, thereby affecting the creation of proteins. In this case, however, people who develop inherited C9orf72-caused ALS and FTD do not carry mutations in exons; instead, the mutations are found in the non-coding DNA segments of C9orf72, called “introns,” that don’t contain instructions for encoding proteins. Non-coding DNA is often dismissed as “junk” DNA, but it can play a role in gene expression.
ALS and FTD patients have hundreds or even thousands of extra sections of nucleotides (the letters of DNA) in the non-coding (intronic) region of C9orf72, repeating the sequence GGGGGCC over and over again. Researchers believe that toxic proteins called dipeptide repeat (DPR) proteins produced by these repeated sequences help drive the neurodegeneration seen in ALS and FTD.
Generating animal models that carry multiple copies of the repeating GGGGCC sequence has been notoriously difficult over the past 10 years, but in a study published recently in Nature Communications, researchers from the University of Chicago did just that. Yoshifumi Sonobe, PhD, a research associate, has been leading a collaborative ALS project between the labs of Raymond Roos, MD, the Marjorie and Robert E. Straus Professor in Neurologic Science, and Paschalis Kratsios, PhD, Assistant Professor of Neurobiology, both senior authors of the new study.

Yoshifumi Sonobe, PhD
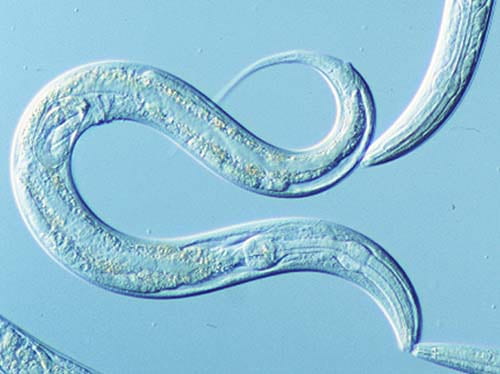
The team turned to a classic scientific tool, the C. elegans roundworm, to create a new model of C9orf72-associated ALS and FTD. Scientists work with C. elegans to study neurodegenerative disorders because they can use powerful genetic techniques with its simple nervous system. C. elegans has just 302 neurons to perform different movement and sensory functions, so researchers can single out different genes easily and track their activity over the short lifetime of the worm (about three to four weeks).
Guided by his earlier work with cultured mammalian cells and chick embryos, Sonobe generated C. elegans strains with 75 GGGGCC repeats. The worms generate the same toxic DPRs, display neurodegeneration and motor defects, and have shorter lifespans than wild type worms, creating an effective tool to understand the mechanisms of mutant C9orf72-caused ALS and FTD.
“The difficult part is the unusual nature of the mutation that causes these forms of ALS and FTD,” Kratsios said. “As you can imagine, it’s very hard to model in an animal. It’s not like having one gene that is mutated in a human and mutating that same gene in a mouse. It’s just a totally different ballgame, and now we have this simple but powerful model of ALS/FTD in C. elegans.”
After further study of the molecular components that make these DPRs toxic to the nervous system, the researchers identified a gene called eIF2D that initiates the process of DPR synthesis. This gene appears to produce the toxic DPRs in an unconventional manner from the non-coding region of DNA. When worms were cross bred to have a mutation that switched the eiF2D gene off, they produced fewer DPRs, their movement improved and they had a longer lifespan, suggesting that eiF2D plays a critical role in the development of disease.
Singling out this mutation in eiF2D is important because that gene is conserved across all eukaryotes, or multicellular organisms. If scientists can recreate such a model in more complex animals that are more like humans, such as mice, they can continue their search for ways to target these genes and mutations for therapy. One promising avenue is working with stem cells taken from human patients with C9orf72-induced ALS that can be coaxed into developing into different types of cells, including neurons. This could reveal more about the more than 30 neurological diseases that are linked to repeating sections of genetic code, including Huntington’s disease and myotonic dystrophy.
“It could be that DPRs and eIF2D may also be important in these other repeat diseases,” Roos said. “Even if we have difficulty with the mouse or stem cell experiments, just to learn that a factor like eiF2D is critically important for disease is an amazing and important discovery.”